臨床成績
国内第Ⅰ/Ⅱa相試験(GD2-PⅠ試験)、国内第Ⅱb相試験(GD2-PⅡ試験)及び海外第Ⅲ相試験(DIV-NB-301試験)は一部国内での承認内容と異なる成績が含まれていますが、承認時評価資料のため掲載します。
海外第Ⅲ相試験(DIV-NB-301試験)
(無作為化、非盲検、多施設共同、並行群間比較試験、海外データ、2001-2012年)
承認時評価資料:海外第Ⅲ相試験(DIV-NB-301試験)
[目 的]
自家幹細胞移植(ASCT)施行前に受けた治療に対してCR*1、VGPR*2又はPR*3を達成した高リスク群神経芽腫患者を対象として、イソトレチノイン*4とユニツキシン免疫療法(ユニツキシン+サルグラモスチム+アルデスロイキン)の併用群[RA+免疫療法群]は、骨髄破壊療法及び自己幹細胞救済療法後の無イベント生存期間(EFS)をRA単独療法群[RA療法群]と比較して改善するか検討する。
*1 CR:完全奏効、*2 VGPR:非常に良い部分奏効、*3 PR:部分奏効、*4 イソトレチノイン:ビタミンA誘導体
[対 象]
有効性解析対象集団:
intent-to-treat(ITT)解析対象集団 226例
(RA+免疫療法群:113例、RA療法群:113例)
安全性解析対象集団:
プロトコール治療を受けた全ての患者240例
(RA+免疫療法群:134例、RA療法群:106例)
[方 法]
以下の通り、ランダムに割り付けを行った。なお、ASCT施行後の生検により残存病変が確認された患者は非ランダムにRA+免疫療法群に割り付けた(Stratum07の患者:ITTからは除外、安全性解析対象集団には含めた)。
<試験デザイン>
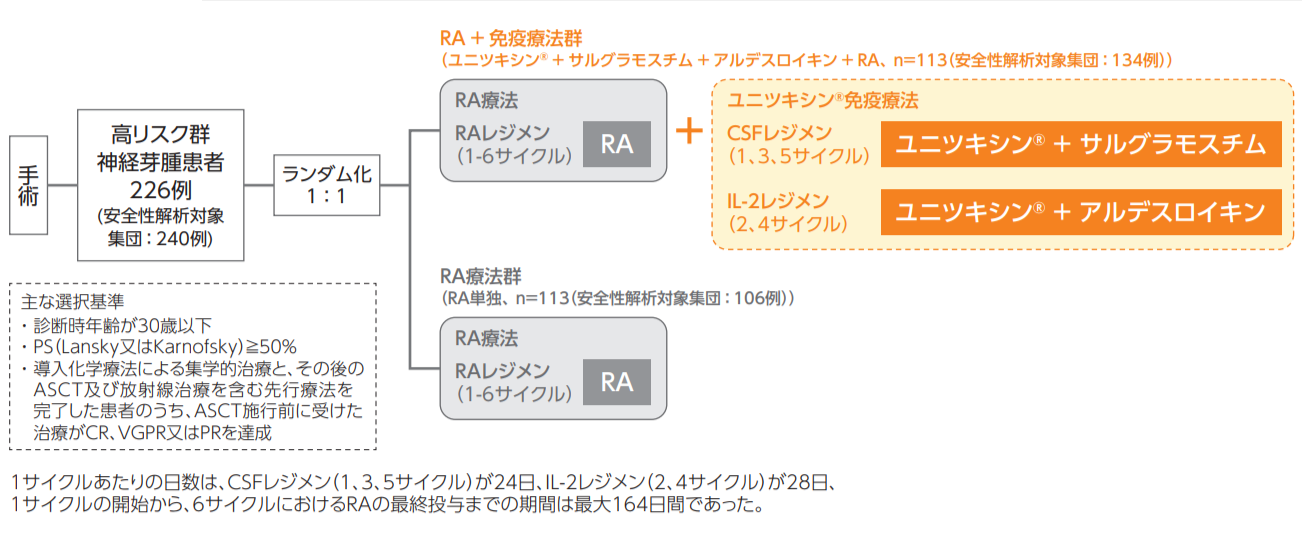
<投与レジメン>
ユニツキシン免疫療法
1-5サイクル
ユニツキシン 25mg/m²を10~20時間かけて静脈内投与。投与は4日間連続で行い、ユニツキシンは4週ごとに投与する。
1、3、5サイクル
サルグラモスチム 250μg/m²を皮下投与(推奨)又は2時間かけて静脈内投与。サルグラモスチム投与は、ユニツキシン投与の3日前から開始して計14日間行う。
2、4サイクル
第1週にアルデスロイキン 3MIU/m²を持続点滴で4日間投与。
第2週にアルデスロイキン 4.5MIU/m²を持続点滴で4日間投与。
RA療法
1-6サイクル
各サイクルの最後の2週間に、RA 160mg/m²(体重12kg超)又は5.33mg/kg(体重12kg以下)を経口投与。
[評価項目]
主要評価項目:
無イベント生存期間(EFS:再発、PD、続発性悪性腫瘍又は死亡をイベントとする)
副次評価項目:
全生存期間(OS)など
※本試験のRA療法群、RA+免疫療法群で使用されているサルグラモスチム、アルデスロイキン、イソトレチノインは国内未承認の薬剤です。
※国内におけるユニツキシン点滴静注17.5mg/5mLの承認された用法及び用量は「フィルグラスチム(遺伝子組換え)及びテセロイキン(遺伝子組換え)との併用において、通常、ジヌツキシマブ(遺伝子組換え)として1日1回17.5mg/m2(体表面積)を10~20時間かけて点滴静注する。28日間を1サイクルとし、1、3、5サイクルは4~7日目、2、4、6サイクルは8~11日目に投与する。」です。
[解析計画]
ITT解析対象集団では、実際に受けた治療に関わらず、ランダム化された治療群に基づいて解析した。RA療法群に割り付けられた患者は、本試験のランダム化部分が早期終了となった後、任意でRA+免疫療法群へのクロスオーバーを行った。有効性の全ての解析において、クロスオーバーを行った患者はランダム化された治療群に基づいて解析し、クロスオーバーを行った時点で打ち切りとした。一方、安全性解析対象集団は、実際に受けた治療(クロスオーバーを行った患者は、クロスオーバー前はRA療法群、クロスオーバー後はRA+免疫療法群)に基づいて解析した。
有効性の主要解析は2009年1月13日カットオフに基づく中間解析とし、安全性解析は2009年6月30日カットオフのデータを用いた。EFS、OSの生存曲線はKaplan-Meier法により推定し、有意水準0.05の両側ログランク検定を用いて、RA+免疫療法群とRA療法群の両群間で差の有無を検討した。
患者背景、ITT解析対象集団
ITT解析対象集団のベースライン時における患者背景は以下の通りであった。
| 項目 | RA+免疫療法群 (n=113) |
RA療法群 (n=113) |
|
| 年齢中央値、歳[範囲] | 3.9 [0.95-15.29] | 3.5 [0.94-13.29] | |
| 性別 | 男性 | 71 (62.8%) | 64 (56.6%) |
| 女性 | 42 (37.2%) | 49 (43.4%) | |
| 民族 | ヒスパニック系又はラテン系 | 11 (9.7%) | 11 (9.7%) |
| 非ヒスパニック系又は 非ラテン系 |
100 (88.5%) | 96 (85.0%) | |
| 不明 | 2 (1.8%) | 6 (5.3%) | |
| 人種 | 白人 | 95 (84.1%) | 90 (79.6%) |
| 黒人又はアフリカ系アメリカ人 | 8 (7.1%) | 8 (7.1%) | |
| アジア人 | 2 (1.8%) | 4 (3.5%) | |
| ハワイ先住民又は その他の太平洋諸島住民 |
0 (0.0%) | 2 (1.8%) | |
| 混血 | 1 (0.9%) | 2 (1.8%) | |
| その他 | 1 (0.9%) | 0 (0.0%) | |
| 不明 | 6 (5.3%) | 7 (6.2%) | |
| ASCT施行前の治療に対する腫瘍縮小効果 | CR | 40 (35.4%) | 38 (33.6%) |
| VGPR | 47 (41.6%) | 49 (43.4%) | |
| PR | 26 (23.0%) | 26 (23.0%) | |
| INSS Stage | Stage 2a | 4 (3.5%) | 0 (0.0%) |
| Stage 3 | 10 (8.8%) | 16 (14.2%) | |
| Stage 4 | 89 (78.8%) | 92 (81.4%) | |
| Stage 4S | 2 (1.8%) | 0 (0.0%) | |
| 欠測 | 8 (7.1%) | 5 (4.4%) | |
| MYCN遺伝子 | 増幅あり | 36 (31.9%) | 45 (39.8%) |
| 増幅なし | 52 (46.0%) | 51 (45.1%) | |
| 欠測 | 25 (22.1%) | 17 (15.0%) | |
| DNA倍数性 | 二倍体 | 35 (31.0%) | 46 (40.7%) |
| 高二倍体 | 49 (43.4%) | 48 (42.5%) | |
| 欠測 | 29 (25.7%) | 19 (16.8%) | |
| 腫瘍細胞の組織型 | 予後良好の組織型 | 4 (3.5%) | 5 (4.4%) |
| 予後不良の組織型 | 68 (60.2%) | 81 (71.7%) | |
| 欠測 | 41 (36.3%) | 27 (23.9%) | |
| 最後のASCT施行からの 期間中央値、日[範囲] | 74 [59-100] | 76 [53-123] | |
| APC中央値、個/μL[範囲] | 2,465 [1,000-369,000] | 2,652 [1,066-550,400] | |
INSS:国際神経芽腫病期分類、APC:絶対食細胞数
非表示部分は左右にスクロールして表示してください。
有効性
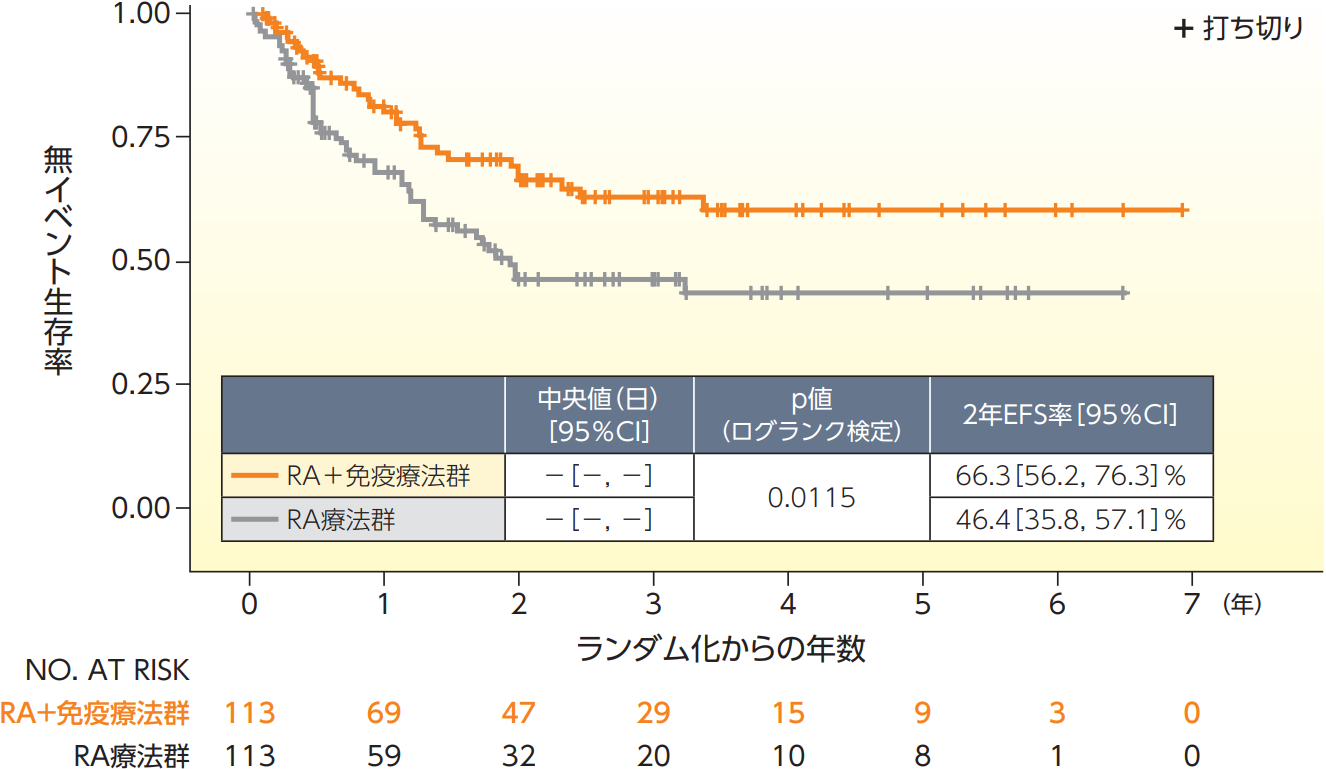
無イベント生存期間(EFS)【主要評価項目、ITT解析対象集団、データカット日:2009年1月13日】
EFSのKaplan-Meier曲線は以下の通りであり、主要有効性解析の時点で打ち切りとなった患者の割合は、 RA+免疫療法群では71%、RA療法群では56%であった。
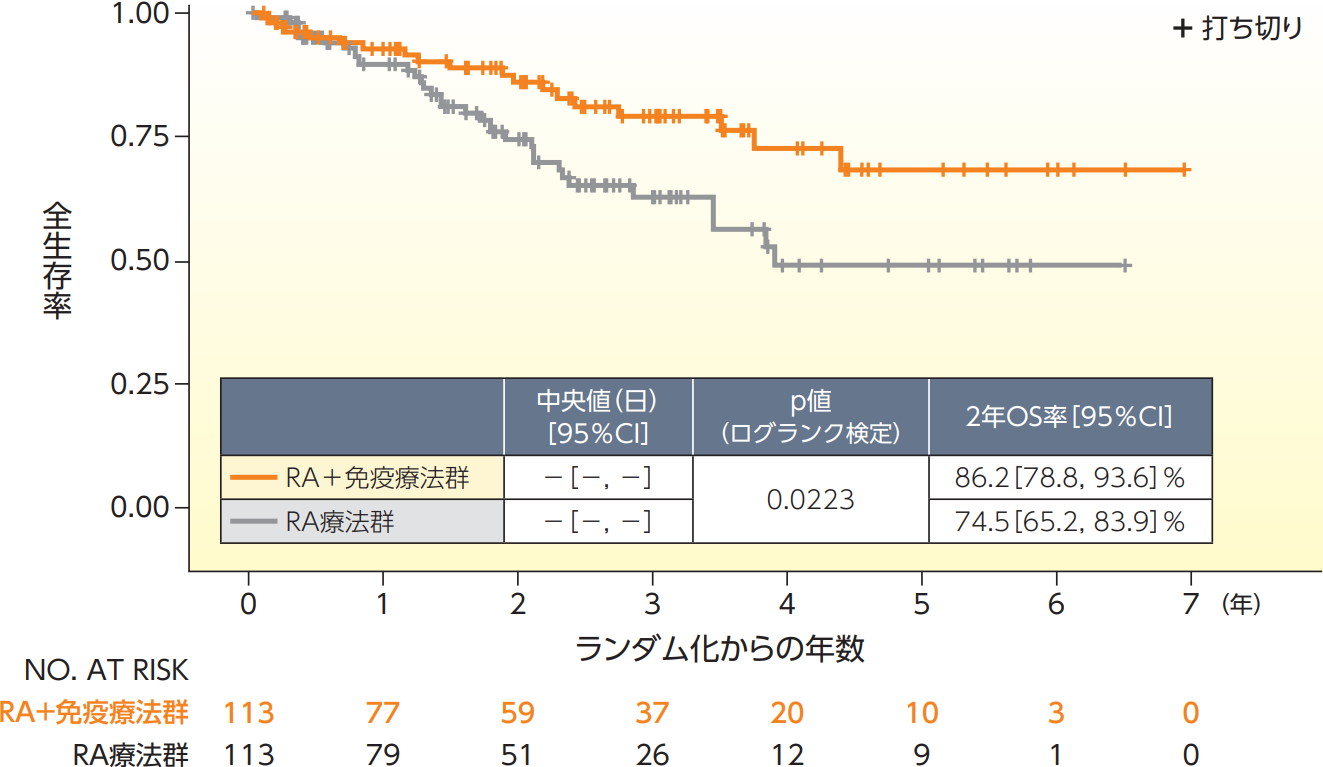
全生存期間(OS)【副次評価項目、ITT解析対象集団、データカット日:2009年1月13日】
OSのKaplan-Meier曲線は以下の通りであり、主要有効性解析の時点で打ち切りとなった患者の割合は、 RA+免疫療法群では83%、RA療法群では71%であった。
安全性
安全性の概要
安全性解析対象集団(RA+免疫療法群134例、RA療法群106例、データカット日:2009年6月30日)のうち、有害事象はRA+免疫療法群132例(98.5%)、RA療法群89例(84.0%)に発現した。重篤な有害事象*1はRA+免疫療法群70例(51%)、RA療法群4例(4%)、投与中止に至った有害事象はRA+免疫療法群6例(4.3%)、RA療法群は不明、死亡に至った有害事象はRA+免疫療法群のみ1例に認められた。
*1 重篤な有害事象:RA+免疫療法群は安全性解析対象集団の134例に加えて、7サイクル目にRA療法群からクロスオーバー4例を含めて138例で解析
<安全性の概要>
| 項目 | RA+免疫療法群 (n=134) |
RA療法群 (n=106) |
| 全事象例数 | 132 (98.5%) | 89 (84.0%) |
| 全事象件数 | 4,734 | 1,026 |
| 治験薬と因果関係がある事象例数 | 131 (97.8%) | 68 (64.2%) |
| 治験薬と因果関係がある事象件数 | 3,962 | 565 |
| 重症度別の事象例数 | ||
| Grade 1 | 108 (80.6%) | 60 (56.6%) |
| Grade 2 | 123 (91.8%) | 57 (53.8%) |
| Grade 3 | 128 (95.5%) | 60 (56.6%) |
| Grade 4 | 73 (54.5%) | 23 (21.7%) |
| Grade 5 | 2 (1.5%) | 0 (0.0%) |
| Grade 3以上の事象例数 | 129 (96.3%) | 66 (62.3%) |
| Grade 3以上の事象件数 | 1,532 | 300 |
CTCAE ver. 3.0
非表示部分は左右にスクロールして表示してください。
<重篤な有害事象、投与中止及び死亡に至った有害事象の内訳>
| 安全性解析対象集団 | RA+免疫療法群 (n=134) |
RA療法群 (n=106) |
| 重篤な有害事象* | 感染症及び寄生虫症17例、 低カリウム血症11例、 低血圧11例、 カテーテル関連感染10例、 アナフィラキシー9例など |
カテーテル関連感染2例など |
| 投与中止に至った 有害事象 |
6例(内訳の詳細な記録なし) | 不明 |
| 死亡に至った 有害事象 |
毛細血管漏出症候群1例 | なし |
MedDRA ver. 13.1
*重篤な有害事象:RA+免疫療法群は安全性解析対象集団の134例に加えて、7サイクル目にRA療法群からクロスオーバー4例を含めて138例で解析
非表示部分は左右にスクロールして表示してください。
主な有害事象(いずれかの群で全Grade50%以上の事象)
| 安全性解析対象集団 | RA+免疫療法群(n=134) | RA療法群(n=106) | ||
| 全Grade | Grade 3以上 | 全Grade | Grade 3以上 | |
| 全事象例数 | 132 (98.5%) | 129 (96.3%) | 89 (84.0%) | 66 (62.3%) |
| 発熱 | 97 (72.4%) | 54 (40.3%) | 30 (28.3%) | 6 (5.7%) |
| 血小板障害 | 89 (66.4%) | 52 (38.8%) | 46 (43.4%) | 26 (24.5%) |
| リンパ球減少症 | 86 (64.2%) | 70 (52.2%) | 39 (36.8%) | 21 (19.8%) |
| 薬物過敏症 | 81 (60.4%) | 35 (26.1%) | 9 (8.5%) | 1 (0.9%) |
| 低血圧 | 80 (59.7%) | 22 (16.4%) | 3 (2.8%) | 0 (0.0%) |
| 低ナトリウム血症 | 77 (57.5%) | 31 (23.1%) | 13 (12.3%) | 4 (3.8%) |
| 腹痛 | 75 (56.0%) | 39 (29.1%) | 9 (8.5%) | 0 (0.0%) |
| アラニン・アミノトランスフェラーゼ増加 | 75 (56.0%) | 31 (23.1%) | 33 (31.1%) | 3 (2.8%) |
| ヘモグロビン* | 68 (50.7%) | 45 (33.6%) | 23 (21.7%) | 17 (16.0%) |
MedDRA ver. 13.1、CTCAE ver. 3.0
*「ヘモグロビン」は、CTCAE ver. 3.0 に基づき、基準値下限未満のヘモグロビン値(すなわち、ヘモグロビン低値)を示す。
非表示部分は左右にスクロールして表示してください。